Example on how to assign CLAYFF atomtypes to a mineral atom struct
See also the advanced examples
Contents
- First set some convenient matlab settings
- Set input and output filenames
- Import a unit cell structure and replicate it into a mineral layer
- Perform isomorphic substitution using substitute_atom
- Assign the CLAYFF (Cygan, 2004) atomtypes to the montmorillonite atom struct
- Write a CLAYFF (Cygan, 2004) .pdb file
- Assign modified CLAYFF atomtypes to the montmorillonite atom struct
- Assign the modified CLAYFF atomtypes to the montmorillonite atom struct
- Heal and assign the modified CLAYFF atomtypes to the montmorillonite atom struct
- Write the new modified CLAYFF .pdb file
First set some convenient matlab settings
format compact; set(gcf,'Visible','on');
Set input and output filenames
filename_in='Pyrophyllite.pdb'; % default is 'Pyrophyllite.pdb' filename_out='6x4x1_MMT_clayff_2004.pdb'; % default is 'Pyrophyllite.pdb'
Import a unit cell structure and replicate it into a mineral layer
Normally, when constructing a mineral particle and assigning the CLAYFF atomtypes, one usually start of by building the mineral particle from an X-ray determined unit cell structure. In this example we will demonstrate how to generate a montmorillonite layer particle from a pyrophyllite unit cell. Both minerals are so-called 2:1 T-O-T sheet silicate minerals, with the difference between the two isostructural minerals being (as you may know) the fact that montmorillonite carries charge defects due to isomorphic substitution. For montmorillonite this means substitution of Si4+ with Al3+ in the two tetrahedral sheets, or octahedral Al3+ with Mg2+ or Fe2+ in the octahedral sheet of the montmorillonite layer.
atom = import_atom(filename_in); % Imports a pyrophyllite unit cell atom = replicate_atom(atom,Box_dim,[6 4 1]); % Replicate the structure by 6x4x1 into a Pyrophyllite clay layer
Perform isomorphic substitution using substitute_atom
substitute_atom was written with centrosymmetric minerals in mind, therfore it works best if the layer is centered at z=0 in the xy-plane. It can handle both substitutions in the octahedral as well as in the tetrahedral sheets. It distributes the substituted sites randomly, except for the fact that one can choose a nearest distance between the substituted sites, which in this example is 5.5 Ångström. We do this to avoid oxygen atoms facing two substituted sites at the same time (Google Löwenstein's rule).
atom = substitute_atom(atom,Box_dim,6*4*2/3,'Al','Mgo',5.5) % Perform octahedral (only) substitutions on 2/3's of all Al sites. Here 5.5 is the minimum distance between the substituted Mg2+ sites % atom = substitute_atom(atom,Box_dim,14,'Al','Mgo',5.5,2,'Si','Al',5.5) % 14 octahedral substitutions and 2 tetrahedral substitutions
Assign the CLAYFF (Cygan, 2004) atomtypes to the montmorillonite atom struct
atom_clayff2004 = clayff_2004_atom(atom,Box_dim,'clayff_2004') % Assign the clayff atom types to the atomstruct % atom_clayff2004 = clayff_2004_atom(atom,Box_dim,'clayff_2004','spc',[1 2]) % In case of 14 octahedral substitutions and 2 tetrahedral substitutions as above, add a final argument [1 2] to iterate over the structure twice the assign the atomtypes correctly
Write a CLAYFF (Cygan, 2004) .pdb file
write_atom_pdb(atom_clayff2004,Box_dim,filename_out); % Print the clay sheet to a .pdb file
Assign modified CLAYFF atomtypes to the montmorillonite atom struct
The original CLAYFF does not contain all different atomtypes needed to model all sorts of clays/minerals, like for instance clay layers with edges. Hence modifications to CLAYFF exist... In this example we are going to use another function, simply called clayff_atom thats different from the original clayff_2004_atom function in two ways. Primarily it uses different atom names (see conversion list below). Secondly, it contains new oxygen atomtypes that do not exist in the original CLAYFF publication from Cygan et al in 2004, which can be used to model lets say clay edges. The partial charges of any new oxygen atomtypes can be derived using the following equation below, and is derived and related to the bond valence of the oxygen atoms, see related references: Tournassat et al, 2016 and Lammers et al, 2017.
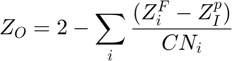
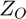 is the partial charge of the oxygen atom, and the summation is taken over all neighbouring cations, where
is the partial charge of the oxygen atom, and the summation is taken over all neighbouring cations, where 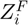 is the formal charge,
is the formal charge, 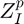 is the partial charge and
is the partial charge and 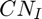 is the coordination number of the cation. Although not explicitly stated/used in the original CLAYFF publication by Cygan et al 2004, this formula can also be used to calculate the partial charge of the oxygen atomtypes in the original CLAYFF. Hence one can assume that the partial charge of any new oxygen atomtype calculated in this way is (more or less...) consisted with all the other atoms/charges in CLAYFF.
is the coordination number of the cation. Although not explicitly stated/used in the original CLAYFF publication by Cygan et al 2004, this formula can also be used to calculate the partial charge of the oxygen atomtypes in the original CLAYFF. Hence one can assume that the partial charge of any new oxygen atomtype calculated in this way is (more or less...) consisted with all the other atoms/charges in CLAYFF.
Below is the list of atomtype names from the Cygan, 2004 paper and the ones modified here, having atomtype names that simply make more sense to me. Note that I have also added a few to the Cygan, (Cygan et al 2004) atomtypes, like oahe/oahhe/oshe etc.
% CLAYFF from Cygan, 2004 = {'h*','ho','o*','oh','ob','obos','obts','obss', 'ohs', 'oas', 'oahhe','oahe', 'oshe','st','ao','at','mgo', 'mgh','cao','cah','feo','lio','Li','Na','K','Rb','Cs','Mg','Ca','Sr','Ba','F','Cl','Br','I'}'; % modified CLAYFF (MHolmboe) = {'Hw', 'H','Ow','Oh','O', 'Omg', 'Oalt','Odsub','Ohmg','Oalsi','Oalhh','Oalh','Osih','Si','Al','Alt','Mgo','Mgh','Cao','Cah','Feo','Lio','Li','Na','K','Rb','Cs','Mg','Ca','Sr','Ba','F','Cl','Br','I'}';
Assign the modified CLAYFF atomtypes to the montmorillonite atom struct
atom_clayff = clayff_atom(atom,Box_dim) % Assign the clayff atom types to the atomstruct % atom_clayff = clayff_atom(atom,Box_dim,'clayff','spc',[1 2]) % In case of 14 octahedral substitutions and 2 tetrahedral substitutions as above, add a final argument [1 2] to iterate over the structure twice the assign the atomtypes correctly
Heal and assign the modified CLAYFF atomtypes to the montmorillonite atom struct
In cases were atoms need healing, or in order to protonate edge groups, one can use a slighlt longer command like below. For more info look into the clayff_atom function and lines 49-80.
atom_clayff = clayff_atom(atom_clayff,Box_dim,'clayff','spc',[1:7]) % Assign the clayff atom types to the atomstruct
Write the new modified CLAYFF .pdb file
write_atom_pdb(atom_clayff,Box_dim,strcat('mod_',filename_out)); % Print the clay layer to a .pdb file